軌道のエネルギー状態は、
- sp3混成軌道
- sp2混成軌道
- sp混成軌道
「結合エネルギー」は、それを切断するために外部から供給されなければならないエネルギーだから、逆に、安定なものほど大きくなる。
従って、
- まず、単結合、二重結合、三重結合、の順に、「結合エネルギー」は大きくなる。
- それだけではなく、同じ単結合、同じ二重結合、・・・、の中でも、その両端の原子の「混成」状態によって、違いが生じる。
p軌道→sp3混成軌道→sp2混成軌道→sp混成軌道→s軌道、の順に、エネルギー状態は低くなるから(これを「混成軌道のs性が高くなる」と言うらしい)、それだけ、「結合エネルギー」は大きくなる。
- [2008前半・1]
- (a)Fsp3・Csp
- (b)Fsp3・Csp2
- (c)Fsp3・Csp3
- [2009前半・1]
まず、(a)、(b)、(c)については、いずれも単結合(σ結合のみ)で、両端の炭素の混成状態を見ると、- (a)Csp3・Csp3
- (b)Csp・Csp3
- (c)Csp2・Csp3
(d)は、Csp2・Csp2で、σ結合のみならずπ結合も持っている。だから、その「結合エネルギー」は、単結合のものより大きい。
従って、(d)>(b)>(c)>(a)。
「結合エネルギー」が大きいほど、結合距離は短い、と思われるから、短い順に、(d)>(b)>(c)>(a)。
ちなみに、2炭素原子感の結合距離では、エタン、ベンゼン、エチレンの順に短くなる、と言われている。これを称して、「ベンゼン環は1.5重結合だ」、などという。ベンゼン、エチレンは、どちらもsp2混成だが、- エチレンでは、2個のπ電子が、2個の炭素原子に、
- ベンゼンでは、6個のπ電子が、6個の炭素原子に、
- [2010前半・1]
これは、わかりません。
どれも、Csp2・Csp2だと思うが、しいて違いを発見するとすると、- (a)では、両端の炭素とも、あと2方向のCsp2・Csp2、
- これに対して、(b)では、左側の炭素は、あと2方向のCsp2・Csp2だが、右側の炭素は、Csp2・Csp2とCsp2・Hsp3
- (c)(d)では、両端の炭素とも、Csp2・Csp2とCsp2・Hsp3。これらに違いが生ずるとすると、10個のπ電子が10個の炭素原子に「非局在化」した電子雲の、対称軸上にある(c)と、偏った場所にある(d)ということになるだろう。
- X-O-Hという構造で、
- Xが金属元素(電気陰性度小)の場合、OはXから十分な電子の供給を得ることができるから、O-H間の電子分布はそれほど偏らず、X+、OH-分離する傾向が高い。つまり「塩基」になる。
- Xが非金属元素(電気陰性度大)の場合、OはXからは十分な電子の供給を得ることができないので、O-H間の電子分布はO側に著しく偏り、電子を失ったHが、H+となって離脱する。つまり「酸」になる。
- 有機化学では、骨格が炭素であるから、「酸」はほぼすべて、C-O-H構造と考えてよい。「アルコールまたはフェノール」、及び「カルボン酸」であるが、アルコールでは、「メチル基の著しい電子供与性」によってH+の離脱が妨げられるから、ほぼ、「酸」ではない、と説明されている。
- 有機化学での「塩基」は、ほぼ、「アミン」しか考えない。アンモニアの水中での電離、
NH3+H2O⇔NH4++OH-
では、- プロトンH+が、H2OからNH3に「供与」されているから、H2Oが「ブレンステッド・ローリー」的には「酸」であり、
- NH3が、その「非共有電子対・孤立電子対・ローンペア」を、NH4+の配位結合のために「供与」したから、「ルイス」的に「塩基」である、
- 「酸」とは、C-O-Hの、「O」の電子状態、
- 「塩基」とは、アミノ基の「N」の「ローンペア」の電子状態、
|
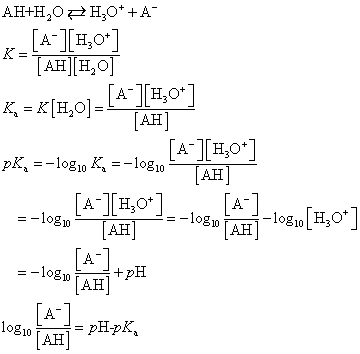
「酸」としての電離平衡定数Kaは、酸の種類に固有な、温度のみに依存する定数である。まわりの環境のpHを指定すれば、この式から、「解離度」が算出できる。
Kaが大きいほど、電離してH3O+を放出できるのだから、強い酸である。従ってpKaが小さいほど、強い酸となる。
- [2008前半・2]
(2)すべて芳香族カルボン酸である。カルボキシル基の接続しているベンゼン環の炭素に対して、「パラ」位に、- (a)-H(水素)
- (b)-Cl(塩素)
- (c)-F(フッ素)
- (d)-CH3(メチル基)
反対に、メチル基は、極めて電子供与性が高い、と言われている。なぜそうなのか?、は、私にはうまく説明できないし、ものの本にも、「実はうまく説明できない」(笑)と書いてあったりもするから、受け入れることにしよう。
- 上の左図のように、ベンゼン環の1位の炭素に、電子吸引性の基が付くと、4位の電子密度は小さくなり、めぐりめぐって4位に接続したカルボキシル基の酸素の電子密度も小さくなる。だから、より強い酸になる。
- 上右左図のように、ベンゼン環の1位の炭素に、電子供与性の基が付くと、4位の電子密度は大きくなり、めぐりめぐって4位に接続したカルボキシル基の酸素の電子密度も大きくなる。だから、より弱い酸になる。
ClとFとでは、Fの方が電気陰性度が大きいから、酸の強さの順位は、
F-C6H4-CO2H>Cl-C6H4-CO2H>C6H4-CO2H>CH3-C6H4-CO2H
「酸の強さの順位」はそのまま「Kaの小さな順位」である。
- [2009前半・2]
- (1)(a)がフェノール、(b)と(d)がカルボン酸、(c)がアルコール。
- フェノール(a)とカルボン酸(b)(d)
- カルボン酸は、O=CR-OHと書いてみれば、電離したとき、O=CR-O-となるけれども、実は、二つの酸素は対等で、本当は、O--CR=Oかも知れない。どっちの酸素が負に帯電しているか?、は断定できない。実際に存在しているのは、これら二つの「極限的な」構造式の確率論的な「重ね合わせ」にあるのだ、というのが、「共鳴」理論。
「共鳴」できるスタイルが多数あればあるほど、エネルギーを放出して「安定化」できる、と言われている。これを「共鳴安定化エネルギー」と呼ぶ。 - それに比べると、電離したフェノールイオンC6H5-O-では、共鳴すべき極限構造式が他にないから、Oの負電荷はそこに固着したまま、それほど安定化できない。
- カルボン酸は、O=CR-OHと書いてみれば、電離したとき、O=CR-O-となるけれども、実は、二つの酸素は対等で、本当は、O--CR=Oかも知れない。どっちの酸素が負に帯電しているか?、は断定できない。実際に存在しているのは、これら二つの「極限的な」構造式の確率論的な「重ね合わせ」にあるのだ、というのが、「共鳴」理論。
- アルコール(c)では、メチル基の電子供与性が異様に強いので、類似のC6H5-CH2-も電子供与性、ヒドロキシル基のOの電子密度が高いから、ほとんど酸とは言えないくらい、弱い。
- 同じ芳香族カルボン酸(b)(d)の中では、パラ位に電子吸引性の基が付いている方が、強い酸になる。ニトロ基NO2-は、炭素より電気陰性度の大きい窒素に、さらにもっと陰性の二つの酸素が結合しているから、電子吸引性はかなり大きい。
O2N-C6H4-CO2H>C6H5-CO2H>C6H5-OH>C6H5-CH2OH> - フェノール(a)とカルボン酸(b)(d)
- (2)芳香族アミン(a)(b)(c)と、脂肪族アミン(d)。
- 芳香族アミンでは、アミノ基の窒素のローンペアが、ベンゼン環のπ電子とともに「非局在化」し、電子密度が低くなる。脂肪族アミンではこのような事情がないから、窒素のローンペアの電子密度は高いまま。従って、脂肪族アミンの方が、芳香族アミンより、強い塩基。
- 同じ芳香族アミンの中では、パラ位に電子吸引性の基があると窒素のローンペアの電子密度は低くなり、反対に、パラ位に電子供与性の基があると、窒素ローンペアのの電子密度は大きくなるであろう。酸の場合と反対に、前者のほうが弱い塩基になるだろう。
C6H11-NH2>H3C-C6H4-NH2>C6H5-NH2>Cl-C6H4-NH2
- (1)(a)がフェノール、(b)と(d)がカルボン酸、(c)がアルコール。
発熱反応A+B→C+D(生成物I)または、A+B→E+F(生成物II)において、「生成物Iの反応熱」<「生成物IIの反応熱」とする。
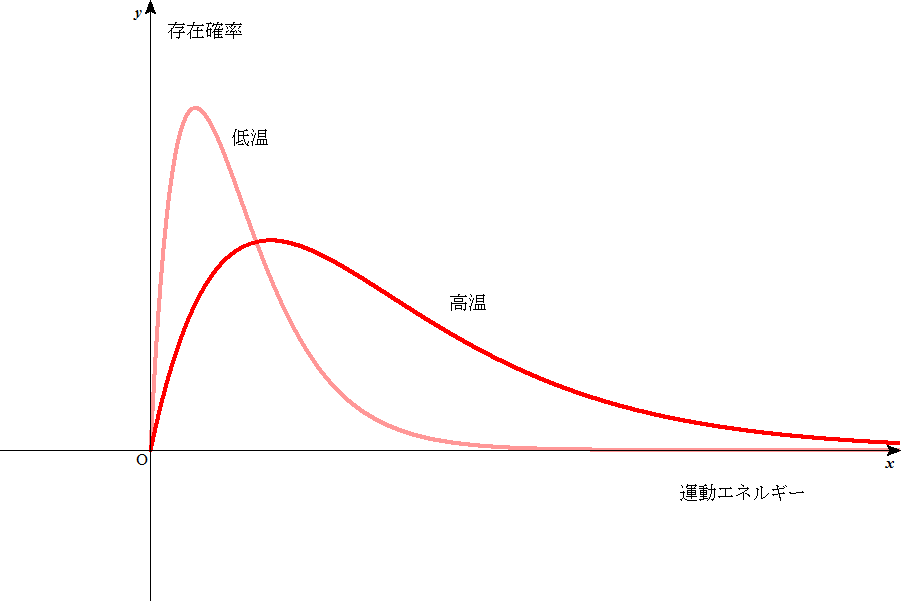 |
| ボルツマン分布(気体分子のエネルギーごとの存在確率) |
このグラフから分かるように、ある「活性化エネルギー」に対して、温度を上昇させると、それを超える分子の存在確率は、増加する。
ここでは必ずしも気体反応、とは限定していないが、溶液中であっても、充分希薄なら、ほぼ同じ事が言えるだろう。
- 「反応Iの活性化エネルギー」>「反応IIの活性化エネルギー」、の場合
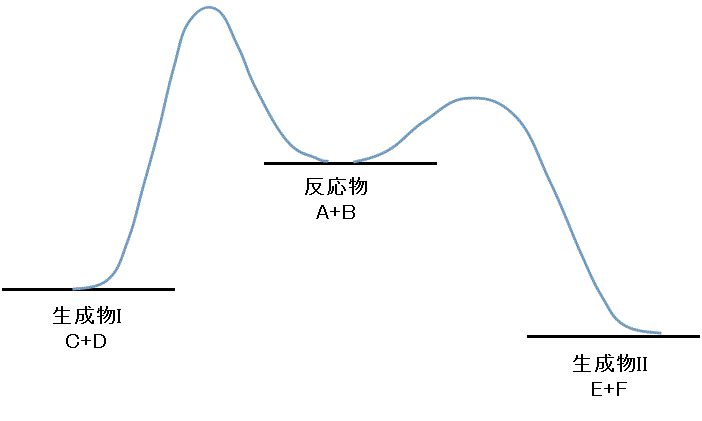
- 低温の条件で反応させたとき
- Iの正反応の活性化エネルギーを超える分子の割合は極めて少ないので、
A+B⇔C+Dは、正反応・逆反応ともに起こりにくい。 - IIの正反応の活性化エネルギーを超える分子の割合は比較的高いので、
A+B→E+Fは、起こりやすいが、
これに比べると、IIの逆反応の活性化エネルギーも結構大きいので、
E+F→A+Bは、あまり起こらない。
- Iの正反応の活性化エネルギーを超える分子の割合は極めて少ないので、
- 高温の条件で反応させたとき
- I、IIの正反応・逆反応いずれの活性化エネルギーも、それを超える分子の割合は増加するが、これらの中で最も低い障壁なのは、IIの正反応の活性化エネルギーなので、
A+B→C+D・・・×
C+D→A+B・・・×
A+B→E+F・・・○
E+F→A+B・・・×
やはり、A+B→E+Fが最も起こりやすく、
- I、IIの正反応・逆反応いずれの活性化エネルギーも、それを超える分子の割合は増加するが、これらの中で最も低い障壁なのは、IIの正反応の活性化エネルギーなので、
- 低温の条件で反応させたとき
- 「反応Iの活性化エネルギー」<「反応IIの活性化エネルギー」、の場合
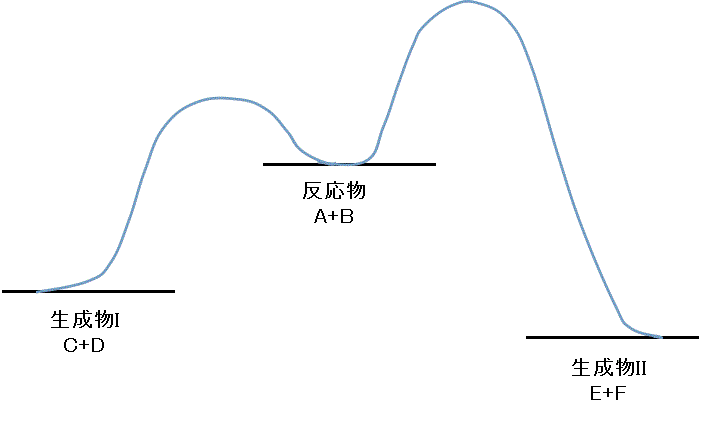
- 低温の条件で反応させたとき
- Iの正反応の活性化エネルギーを超える分子の割合は比較的高いので、
A+B→C+Dは、起こりやすいが、
これに比べると、Iの逆反応の活性化エネルギーも結構大きいので、
C+D→A+Bは、あまり起こらない。 - IIの正反応の活性化エネルギーを超える分子の割合は極めて少ないので、
A+B⇔E+Fは、正反応・逆反応ともに起こりにくい。
- Iの正反応の活性化エネルギーを超える分子の割合は比較的高いので、
- 高温の条件で反応させたとき
- I、IIの正反応・逆反応いずれの活性化エネルギーも、それを超える分子の割合は増加するが、これらの中で最も低い障壁なのは、Iの正反応の活性化エネルギーなので、反応の起こりやすさを評価すると、次のようになるだろう。
A+B→C+D・・・○
C+D→A+B・・・△
A+B→E+F・・・△
E+F→A+B・・・× - Iの正反応は活発に行われるが、同時にIIの逆反応も起こりやすくなるので、これらはやがて「平衡」に達し、もはや、生成物IIの量は増えなくなるだろう。ここで「長時間」という条件を付加しているのはそういうことで、Iが平衡に達した後も、IIは依然として正反応が優勢であるから、十分時間が経過すれば、次第に生成物IIの収量は増加していく。
- I、IIの正反応・逆反応いずれの活性化エネルギーも、それを超える分子の割合は増加するが、これらの中で最も低い障壁なのは、Iの正反応の活性化エネルギーなので、反応の起こりやすさを評価すると、次のようになるだろう。
- 低温の条件で反応させたとき
- 「反応Iの活性化エネルギー」=「反応IIの活性化エネルギー」、の場合
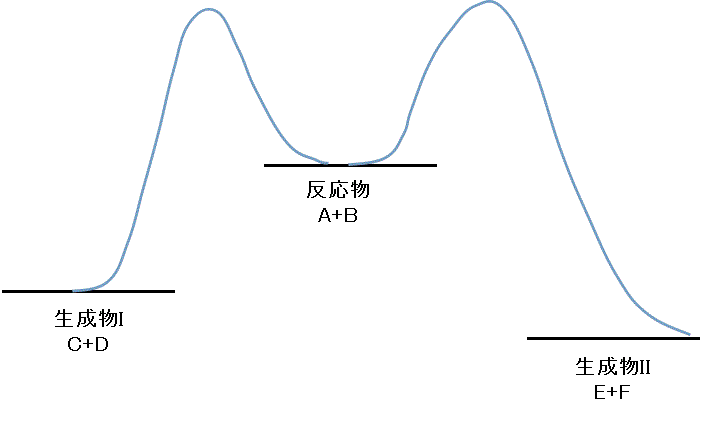
- 低温の条件で反応させたとき
活性化エネルギーの障壁は対等だから、I、IIの正反応はともに同程度に進行し、生成物もほぼ同量生じるだろう。 - 高温の条件で反応させたとき
逆反応の活性化エネルギーは、IIよりIの方が小さいから、Iのほうが先に平衡に達するだろう。Iが平衡に達してもなおIIは正反応の方が優位だから、時間経過とともに生成物IIの割合が増大してくることになるだろう。
低温で短時間の反応条件を採用すると、さしあたり活性化エネルギーの障壁の小さい方の反応が優位になることを、「短期間なら、反応速度の大きいほうが有利」という意味で「速度論支配」と呼んでいるらしい。
反対に、必ずしも高温でなくとも良いのだが、十分に長い時間で考えると、究極的には、最も安定な、・・・、これは、反応熱だけでは決まらないはずで、エネルギー的(熱力学第一法則)にも、エントロピー的(熱力学第二法則)にも、最も安定な状態が選ばれるはずだ、という意味で「熱力学支配」と呼んでいるらしい。
- [2008前半・5]
- 低温・短時間なら、活性化エネルギーの障壁の小さな方が主生成物となる。α-置換体が生成物Iにあたる。
- 高温で、長時間になるほど、生成物II、すなわち、β置換体の収量が増えてくる。
- 低温の条件で反応させたとき
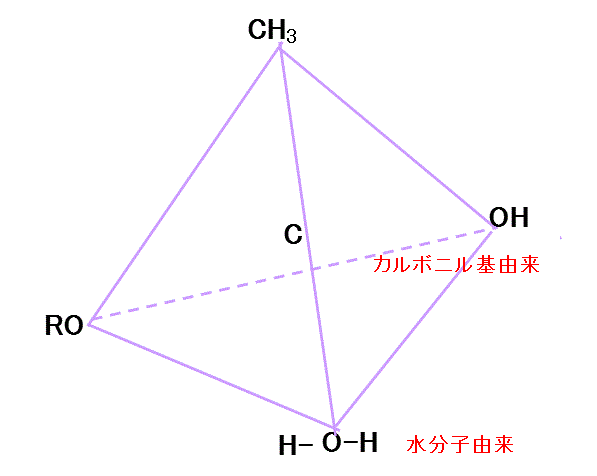
- エステルの塩基による加水分解
- カルボニル基のC=Oのシグマ結合は、C(δ+)=O(δ-)に分極、π電子もO側に偏っている。Cの電子密度は低くなり、正電荷を持つ。
- その正に帯電したカルボニル基のCに、まわりに存在する塩基由来のOH-が接近し、Oの非共有電子対を与える。
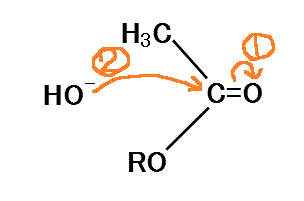
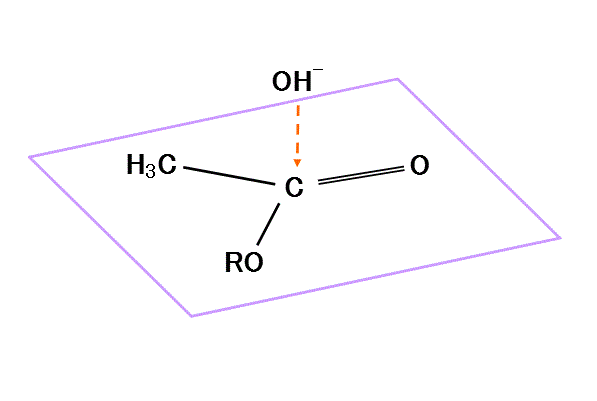
- カルボニル基のCの電子不足状態は解消、カルボニル基のOの過剰な電子は、Cに戻り(ここがよくわからない!、再びπ結合が形成される、ということか?)
- さらにRO-のOに与えられる。十分な電子の供給を得たRO-は、もはやCからの電子供給を受けなくてもよいから、陰イオンとして分離する。
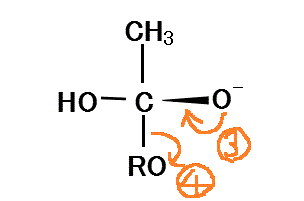
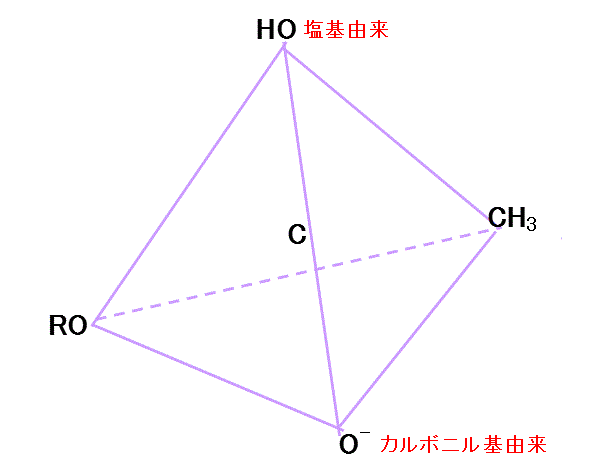
- RO-は隣接する(塩基由来の)-OHのHに非共有電子対を与え、
- それによってOH間の共有電子対はO側に偏り、HはH+として離脱する。
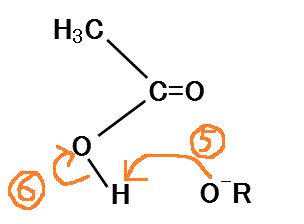
こうして、H3C-C=O-O-+ROHとなる。
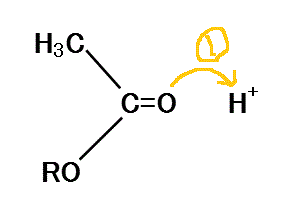
- エステルの酸による加水分解
- 負に帯電したカルボニル基のOが、まわりの酸由来のH+(またはHClなどの酸の分子)に非共有電子対を与える。
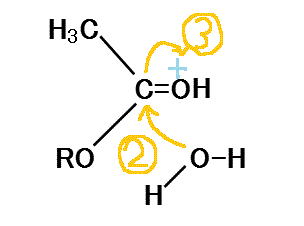
- 正に帯電したカルボニル基のCに、水分子が近づきOの非共有電子対を与える。
- H+を受け取って電子不足になったカルボニル基のOに、カルボニル基のCから電子が流入する。
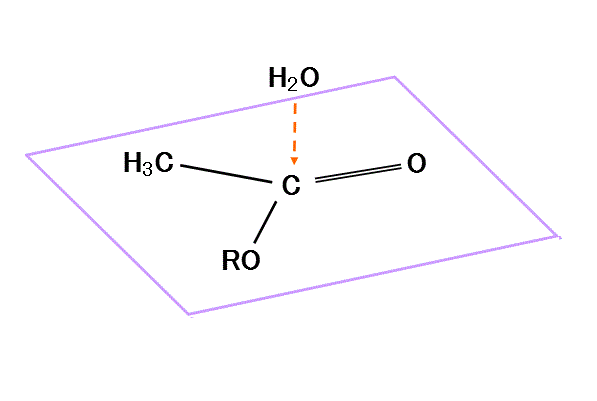
- 水分子の一方のHは、隣接するROのOから、非共有電子対を与えられ、
- このためOH間の共有電子対は、十分O側に偏り、このHは離脱しやすくなる。
こうしてROHが離脱する。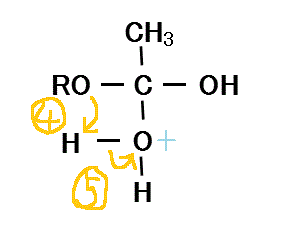
- ROH離脱の際にO-C間の共有電子対は、ROHがもっていったから、Cは正に帯電。残りのO-H結合がこの電子を補い、従ってHがH+として離脱しやすくなる。
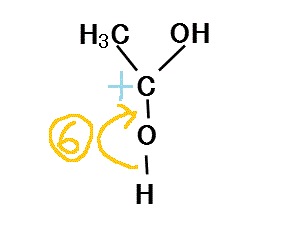
H+が離脱し、CH3-CO-OHが出来上がる。
- 負に帯電したカルボニル基のOが、まわりの酸由来のH+(またはHClなどの酸の分子)に非共有電子対を与える。
- 酸触媒によるエステル化
- 負に帯電したカルボニル基のOが、まわりの酸由来のH+(またはHClなどの酸の分子)に非共有電子対を与える。
- H+を受け取ったことで、カルボニル基のOの電子密度は低くなる。これをCが補う。
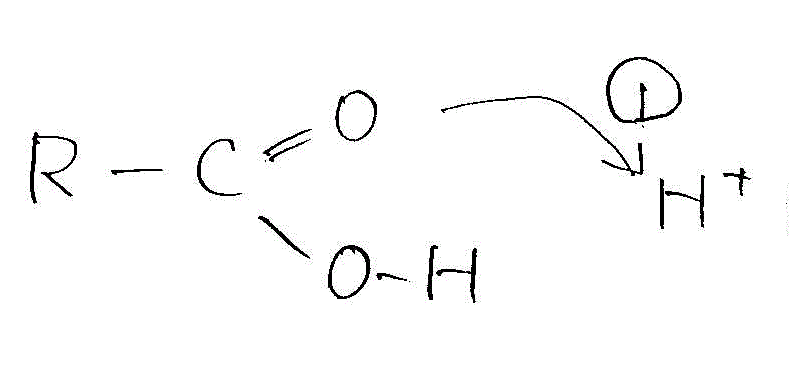 →
→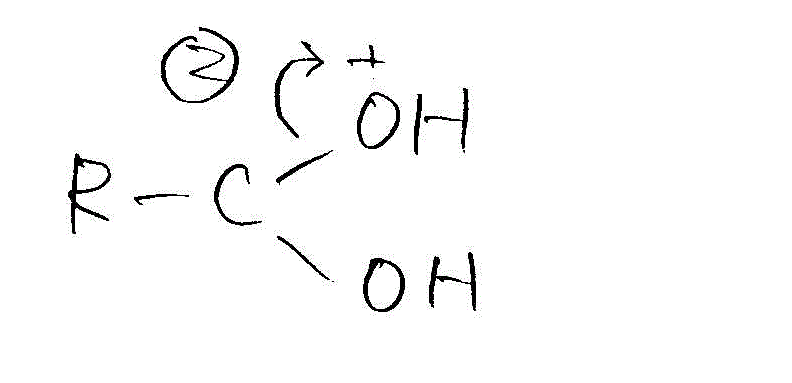
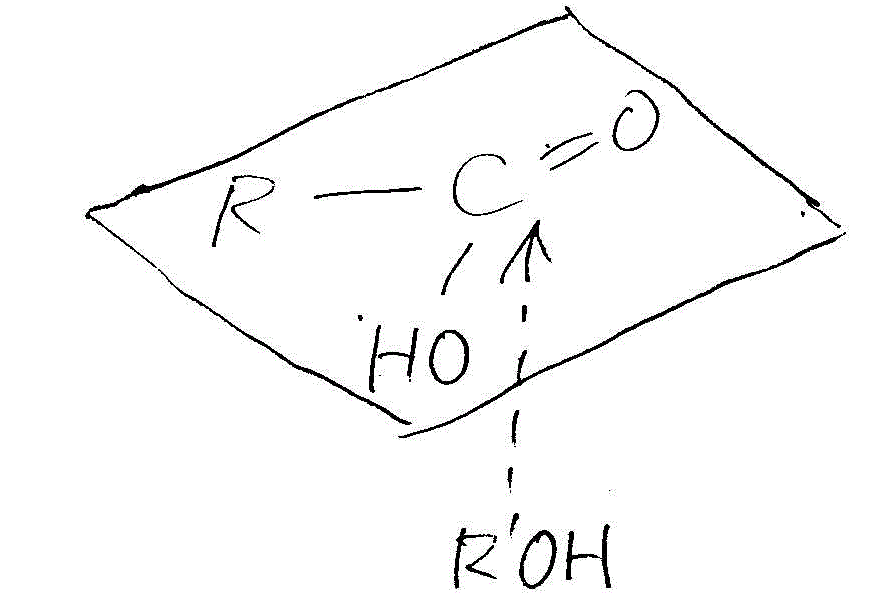
- こうして電子が不足して性に帯電したCに、アルコールのOが非共有電子対を与える。
- アルコールのヒドロキシル基のHは、正に帯電しているから、近くの(どちらだろう?、カルボキシル基のOH由来なのか、カルボキシル基のC=OのO由来なのか?、どちらでも距離はほぼ同じだろう?)OHのOがこれに非共有電子対を与える。
- それによって電子不足となったOはC-O間の共有電子対を引き寄せる。従って、この結合は切れやすくなる。
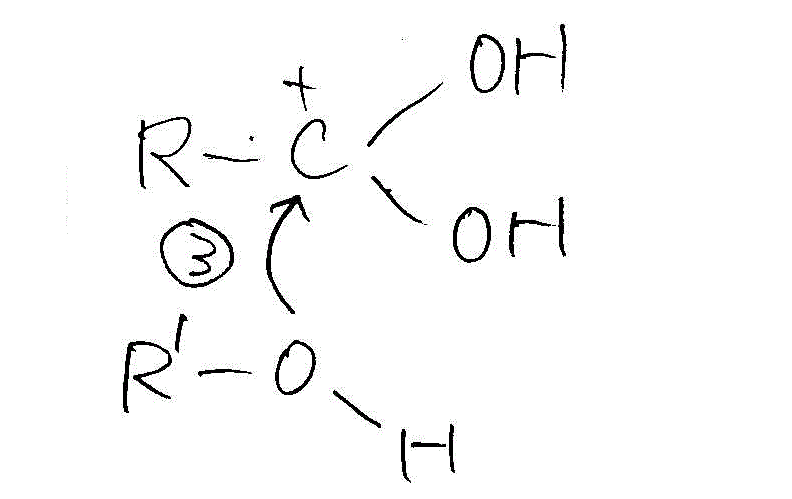 →
→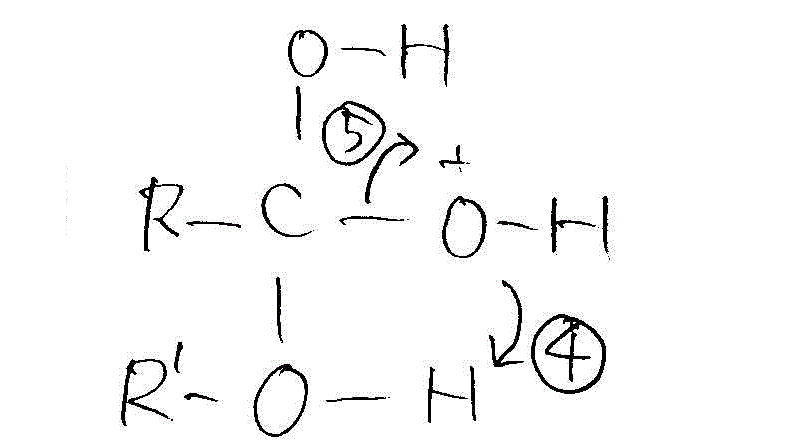

- こうして水分子が離脱する。確かに、カルボン酸からOH、アルコールからHが取れている!
水分子離脱の際に、共有電子対を持って行かれたCは正に帯電しているから、OH間の共有電子対はO側に引き寄せられ、H+が離脱しやすくなる。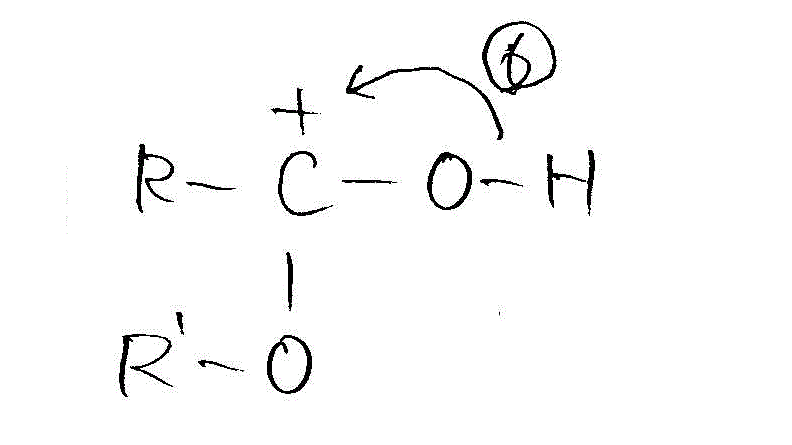 →
→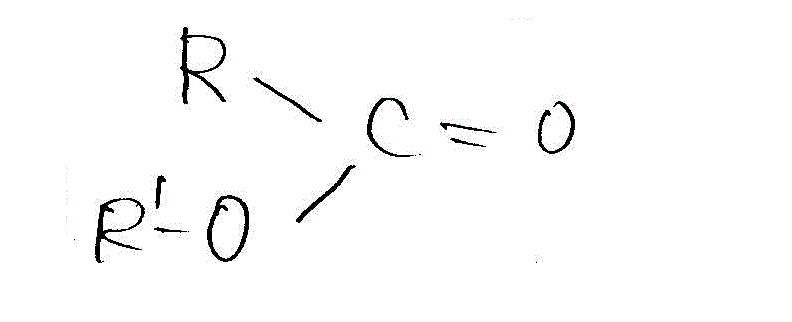
(ちなみに、上の話で、アルコールのヒドロキシル基のHを引き寄せるのがどちらのOなのかによって、中間体のsp3の四面体は、光学異性体の関係になり、生成物のエステルのカルボニル基のOが、はたまた離脱した水分子のOがどちらに由来するものなのかが、異なってくることになる。放射性酸素による標識などで、実験的には、きっと明らかなのだろう。手元の教科書では、上の図のように、カルボキシル基のOH由来のものであるような図が描かれていたから、きっと、そうなのだろう。)
- [2008前半・3](1)
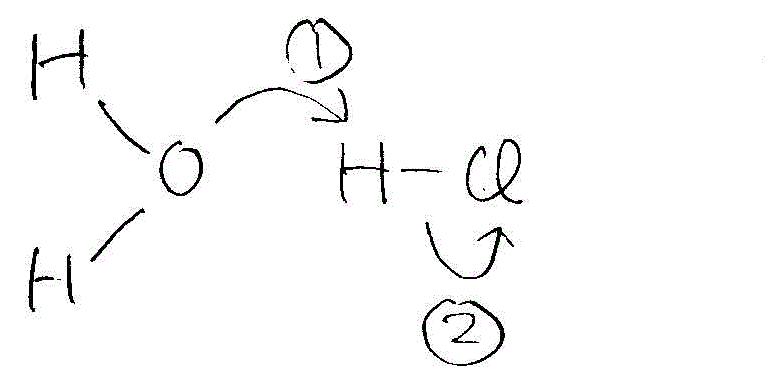 →
→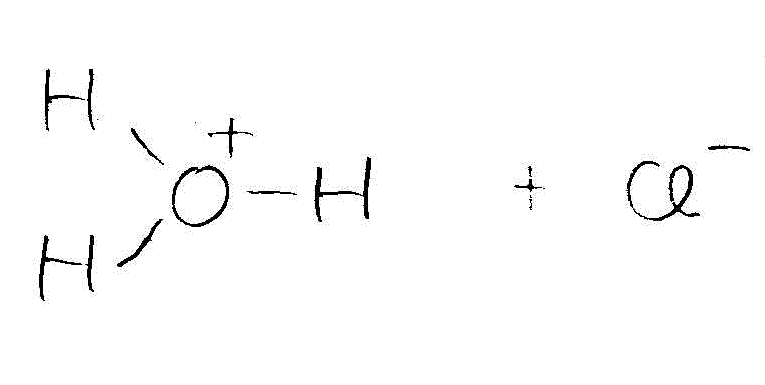
(2)エステルの酸による加水分解。
- [2010前半・5](1)準備中(笑)、(2)エステルの生成(酸触媒によるエステル化)。
- [2011前半・5](1)エステルの塩基による加水分解、(2)準備中(笑)。
- [2009前半・4]濃硫酸存在下の、エタノールの脱水
- それぞれの反応の反応熱を算出する。
- 2H3C-CH2-OH(液)=H3C-CH2-O-CH2--CH3(液)+H2O(液)+Q1
2molのエタノールから脱水して1molのジエチルエーテルができる。Q1=2×234-253=+215
1molのエタノールから脱水して1/2molのジエチルエーテルなら、215÷2=+107.5 - H3C-CH2-OH(液)=H2C=CH2(気)+H2O(液)+Q2
1molのエタノールから脱水して1molのエチレンができる。Q2=234+52=+286
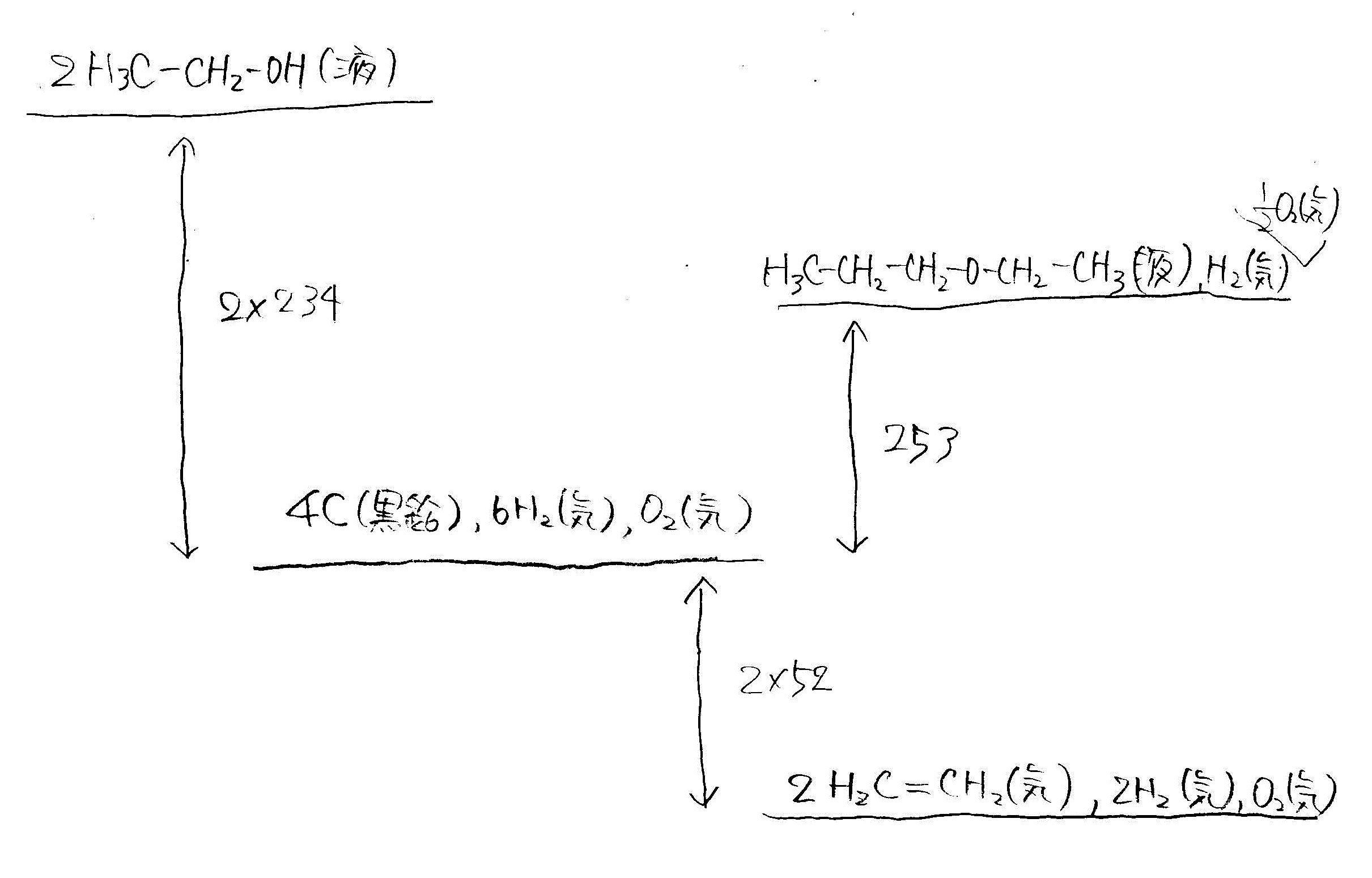
従って、「反応物」エタノール、から「生成物I」ジエチルエーテル、または「生成物II」エチレンを得る反応はいずれも発熱で、3物質のもつエネルギー(自由エネルギー)は、エタノール、ジエチルエーテル、エチレン、の順に低くなる。 - 2H3C-CH2-OH(液)=H3C-CH2-O-CH2--CH3(液)+H2O(液)+Q1
- ところで、実験的には、低温の条件ではジエチルエーテルが、高温の条件ではエチレンが、それぞれ主生成物となることが知られている。
- 低温で短時間の反応では、活性化エネルギーの障壁が低いIの正反応がもっぱら進行し、「生成物I」が主に得られ、
- 高温で長時間反応を続けると、Iは正反応、逆反応ともに活発となって平衡に達し、収量は増えなくなり、やがて「生成物II」が卓越してくることになる。
- 反応機構
- 高温条件(エチレン生成)
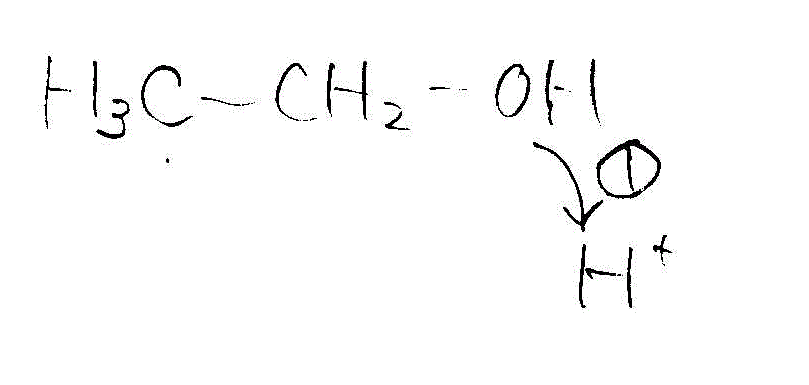 →
→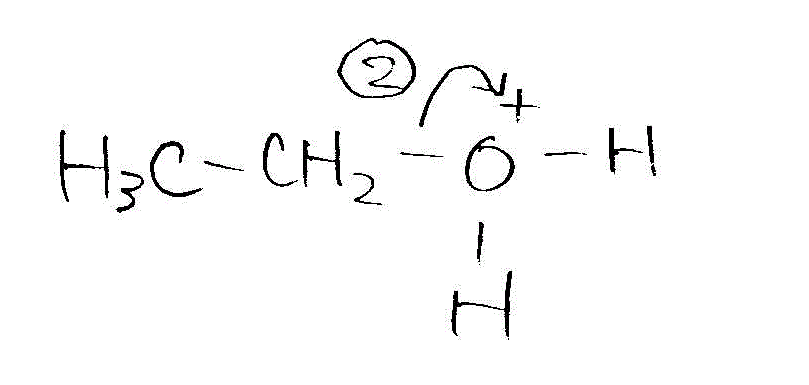 →
→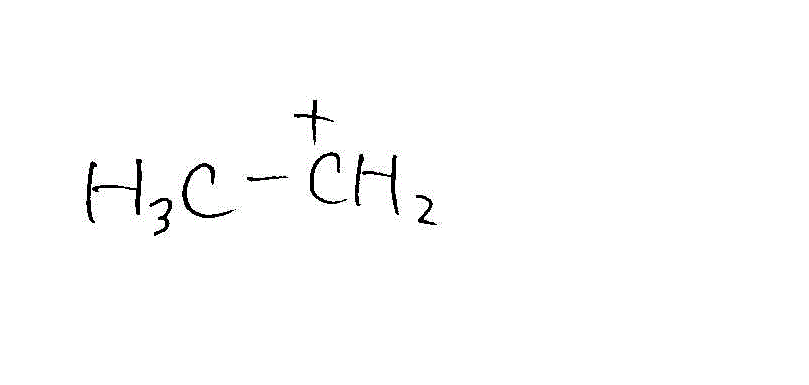 →
→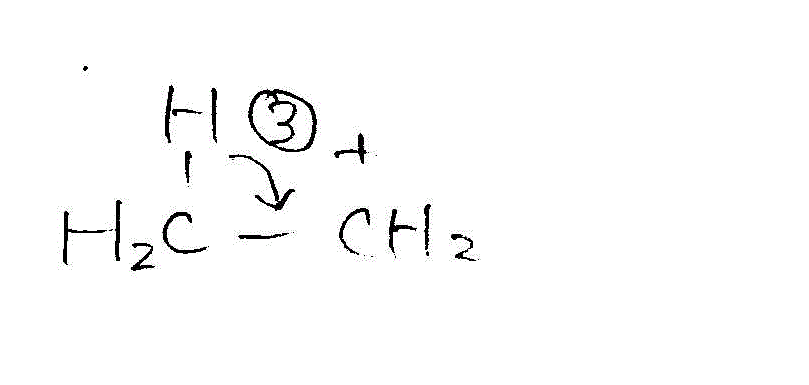 →
→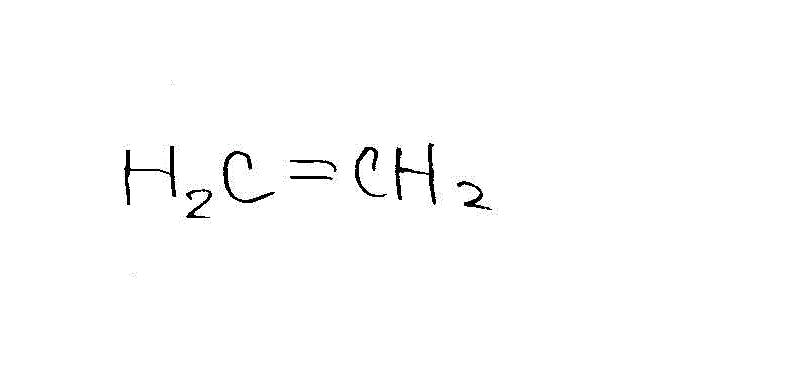
- 低温条件(ジエチルエーテル生成)
→→
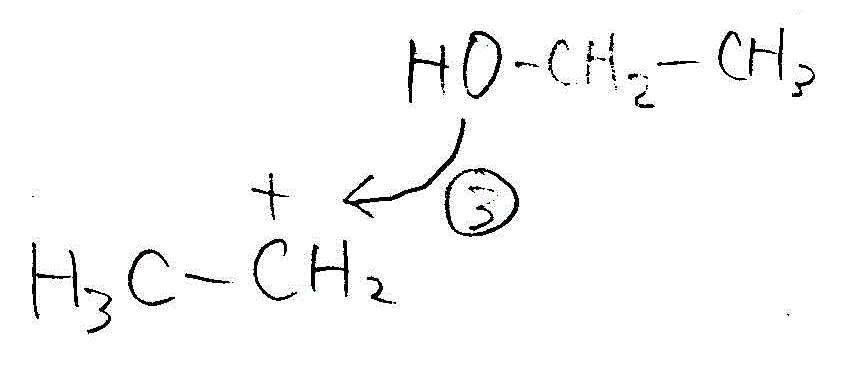 →
→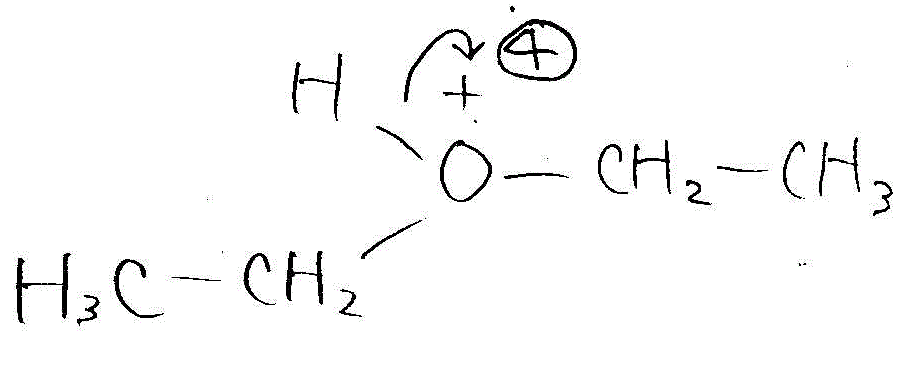 →
→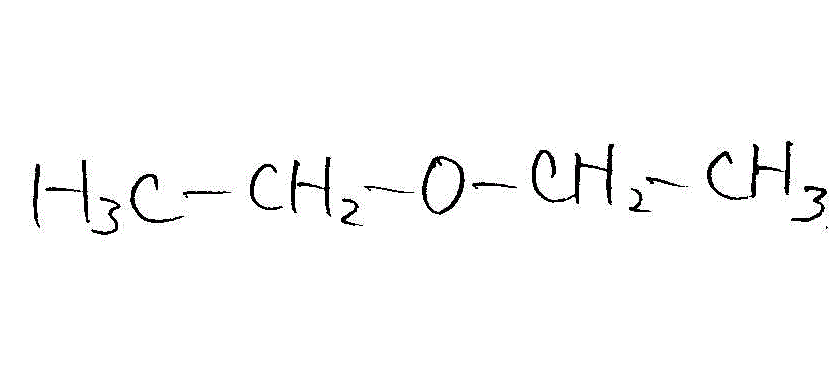
- ジエチルエーテルからエタノールへの分解(酸性下で水と反応)
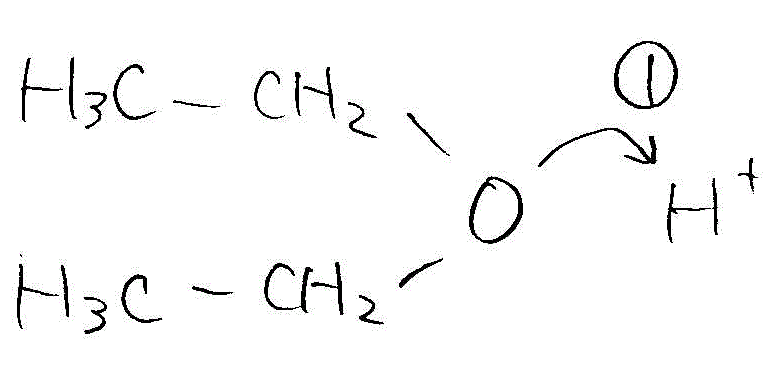 →
→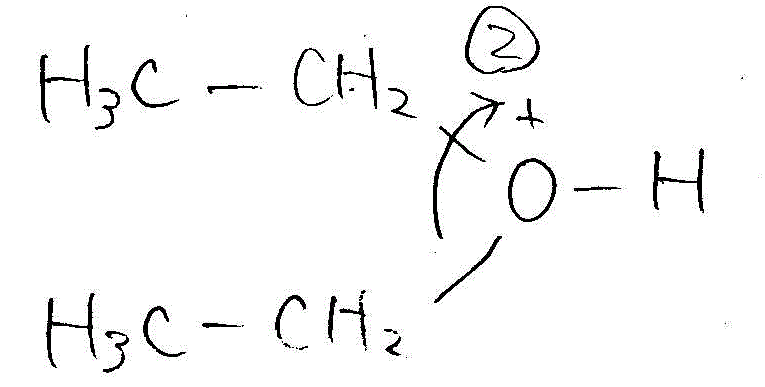 →
→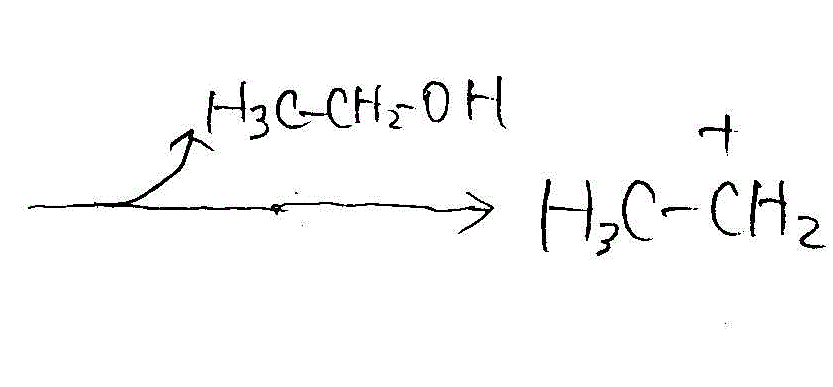
 →
→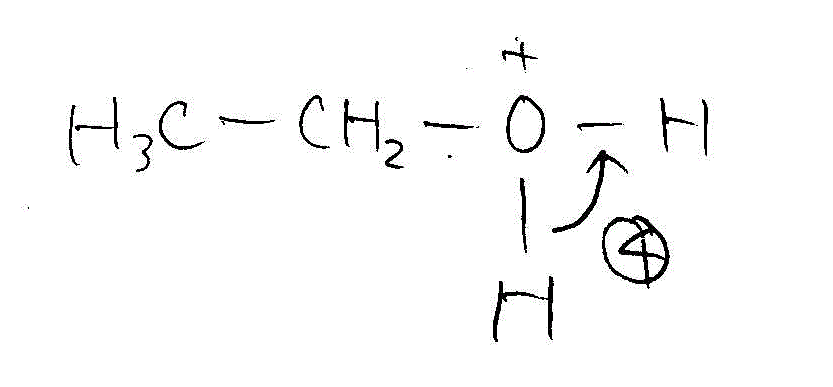 →
→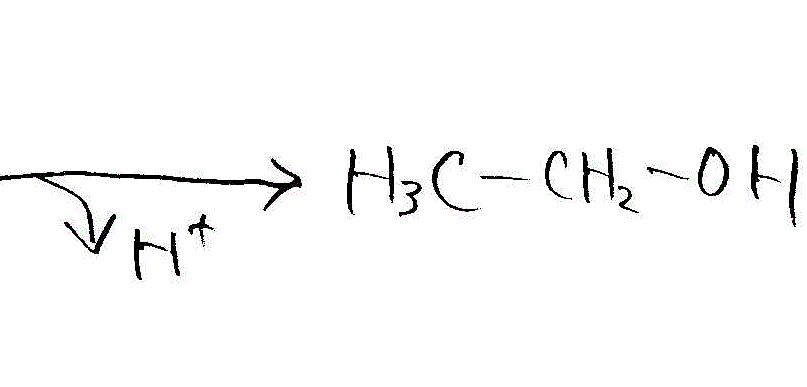
- 高温条件(エチレン生成)
- それぞれの反応の反応熱を算出する。
- 直鎖アルカン
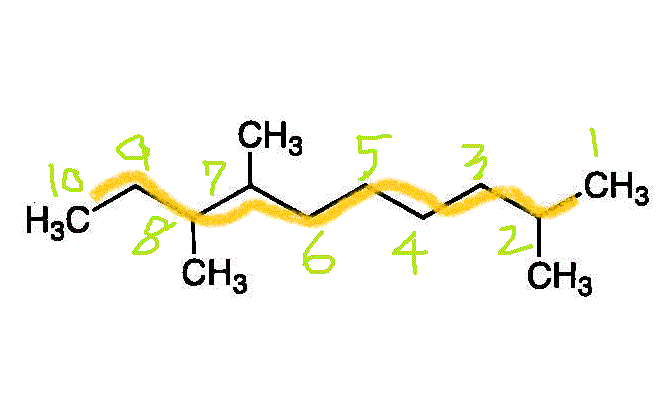
- 一番長い鎖(主鎖)を見つけて、端から番号を付ける。枝分かれの番号が、小さくなるようにする。逆から数えたら3,4,9になるから、こちらを採用する。
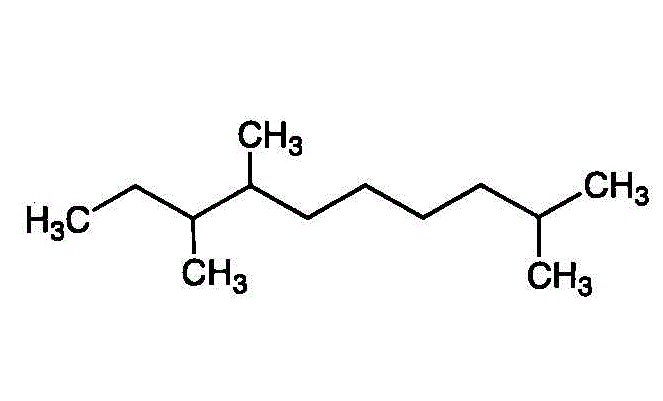
2番、7番、8番に「メチル基」(炭素数1のアルキル基)が付いた、「デカン」(炭素数10のアルカン)・・・2,7,8-trimethyldecane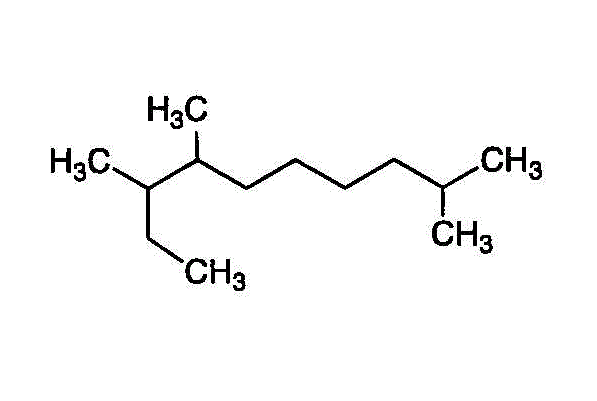
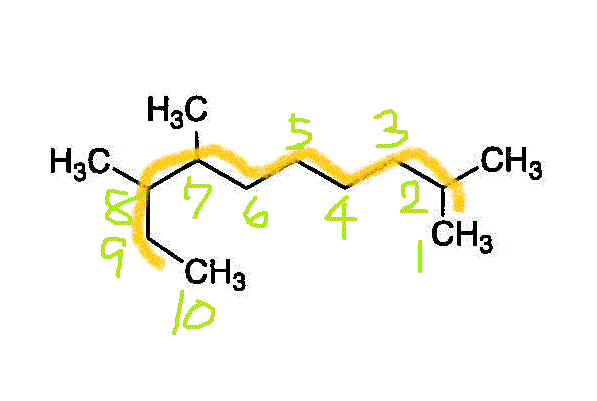
- 2,7,8-trimethyldecane
- 枝分かれには、根元から枝先に向かってから順に番号を付け、括弧書きで表す。
3番には「メチル基」、4番には、枝分かれの1番に「メチル基」が付いた「エチル基」、が付いた炭素数7のアルカン・・・3-methyl-4(1-methylethyl)heptane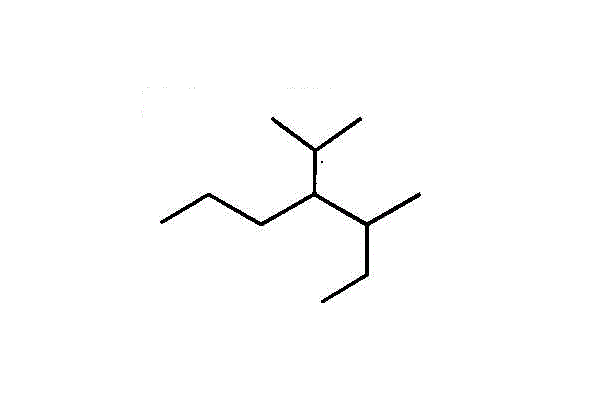

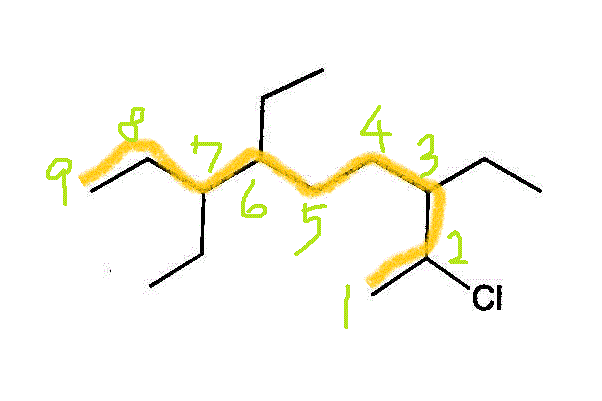
- 異なる置換基があるときは、アルファベット順に各順を決める。「塩素」chloro-と「エチル基」ethyl-では、「c」が先。(端から数えて同じ番号に異なる置換基がある場合も、「主鎖」の番号もこれで決める。ここでは同じだが)。
2番に「塩素」、3番、6番、7番に「エチル基」の付いた、炭素数9のアルカン・・・2-chloro,3,6,7-triethylnonan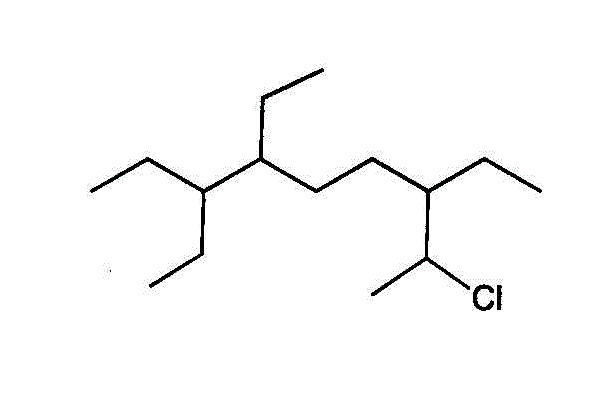
- 反対側から数えたら、3,5,6になる。だから、こちらから数える。・・・3,4,6-trimethyloctane

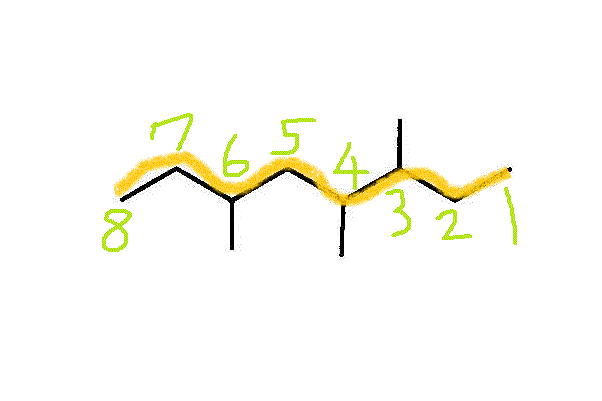
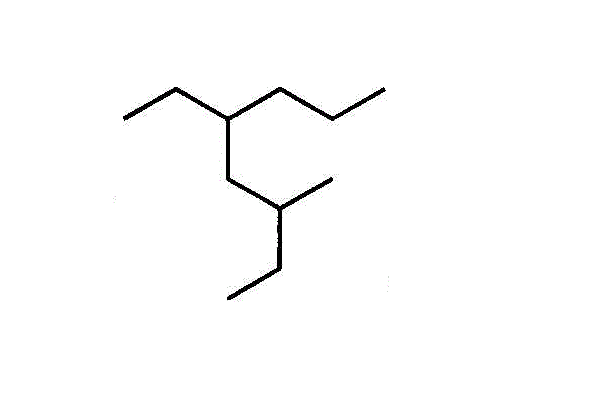
- 3-methyl,5-ethyloctane
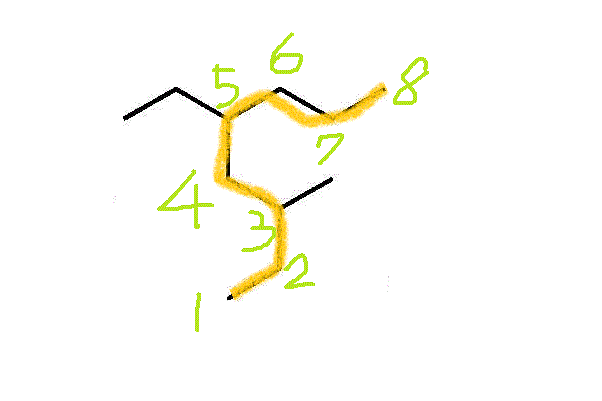
- どちらの端から数えても、4番目に置換基があるが、「塩素の付いたメチル基」chloromethyl-と、「メチル基」methyl-では、アルファベット順に「c」が優先するから、この数え方になる、多分。4番に「塩素のついたメチル基」、7番、8番に2個の「メチル基」の付いた、炭素数11のアルカン(ウンデカン、decaが10、unが1)・・・4-chloromethyl-7,8-dimethylundecane
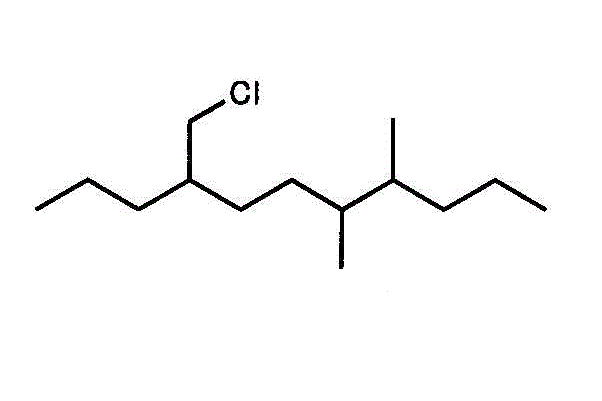
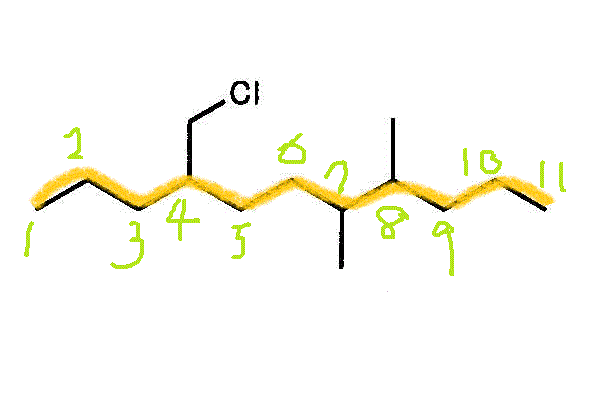
- 一番長い鎖(主鎖)を見つけて、端から番号を付ける。枝分かれの番号が、小さくなるようにする。逆から数えたら3,4,9になるから、こちらを採用する。
- シクロヘキサン2置換体のcis/trans問題
sp3混成の単結合は、理想的にはメタン型正四面体構造、結合角約109°(cosθ=-1/3)をとる。環状構造の場合も、なるべくこれに近い結合角になったとき、安定化する。シクロヘキサンの場合、それが、「椅子型」、「舟形」のどちらか。
2置換体の相互関係は、この歪んだ六角形を「平面」に見立てて、お互い同士が「同じ側」にあれば、「シス」cis、「反対側」にあれば、「トランス」trans。下図では、右のオレンジ色●と、左のブルー●が「シス」、右のオレンジ色●と、左のオレンジ●が「トランス」。
エネルギーを加えてねじると、相互変換を起こすが、両者の相対的な関係は変わっていないことがわかる。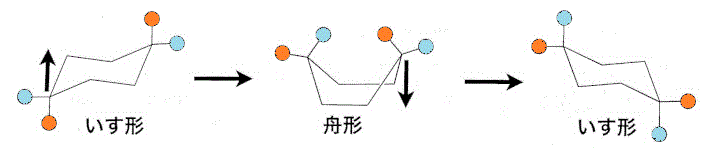
「椅子型」構造の、この六角形の「平面」を赤道面(equator)に見立てて、それに垂直な地球の回転軸(axis)方向をaxial「アクシアル」、赤道面から外向き方向をequatorial「エクアトリカル」、と呼ぶ。上の左端の図の「椅子型」では、オレンジ色●が「アクシアル」、ブルー●が「エクアトリカル」なのに、左端を持ち上げて「舟形」にし、次に右端を押し下げて別の上の右端の図のような「椅子型」にすると、今度は、ブルー●が「アクシアル」、オレンジ●が「エクアトリアル」、と反転している。- 六角形の「面」の「上側」をオレンジ●、「下側」をブルー●で表すと、2つのメチル基は、「反対側」、だから「トランス」。・・・trans-1,2-dimethylcyclohexan
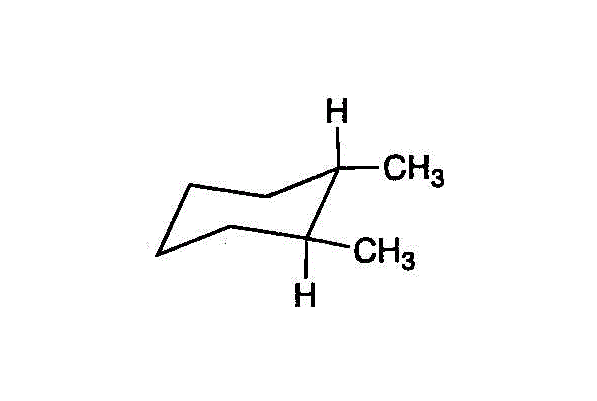
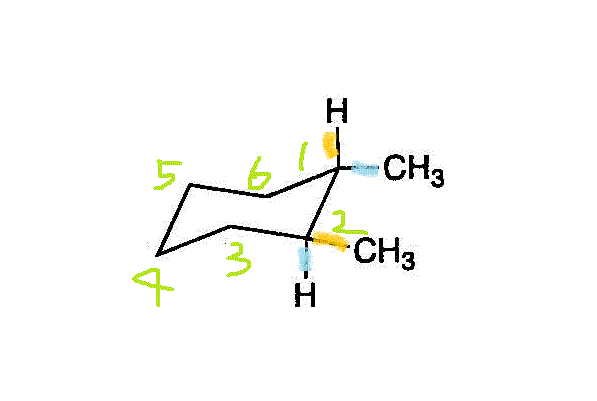
- 省略されている-Hを書き込んでみると、・・・cis-1,2-dimethylcyclohexan

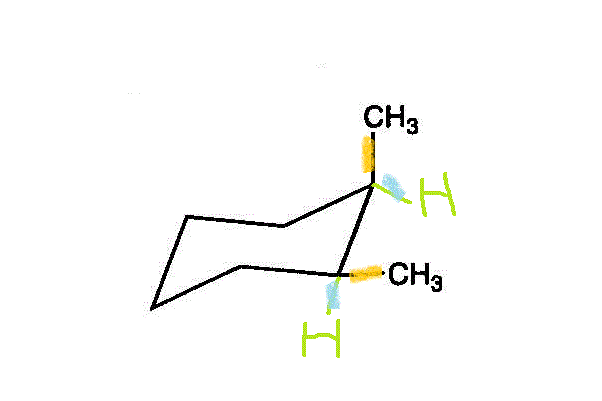
- ・・・trans-1,3-dimethylcyclohexan
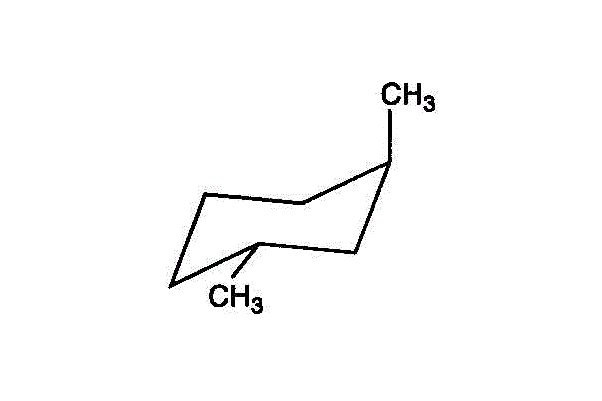
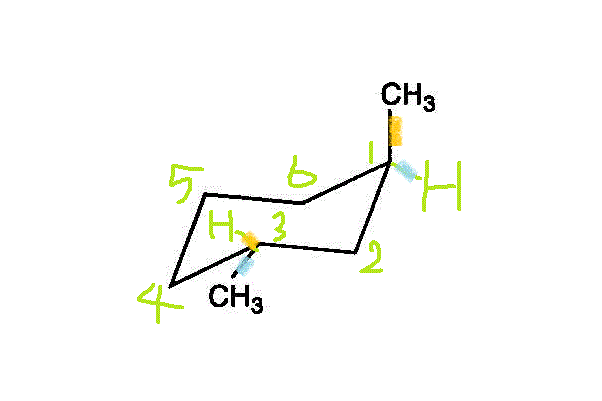
- ・・・trans-1,4-dimethylcyclohexan
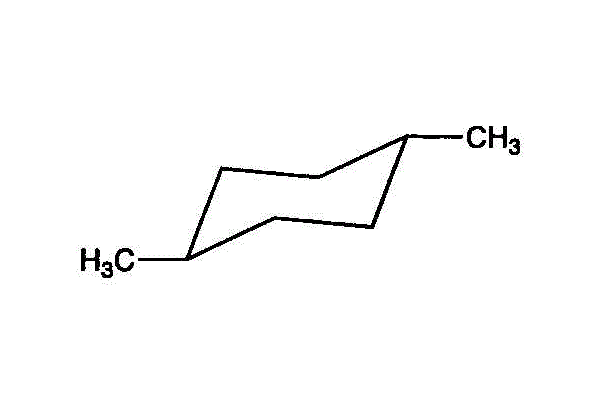
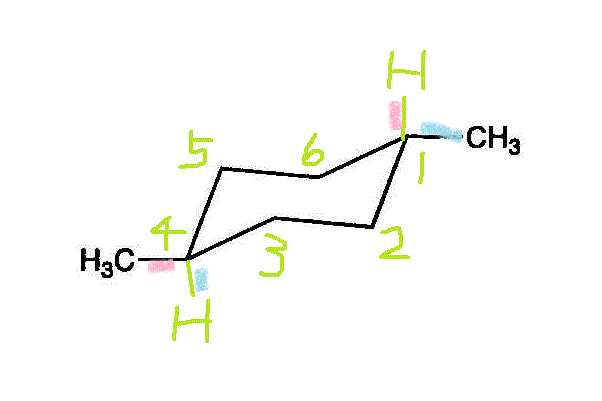
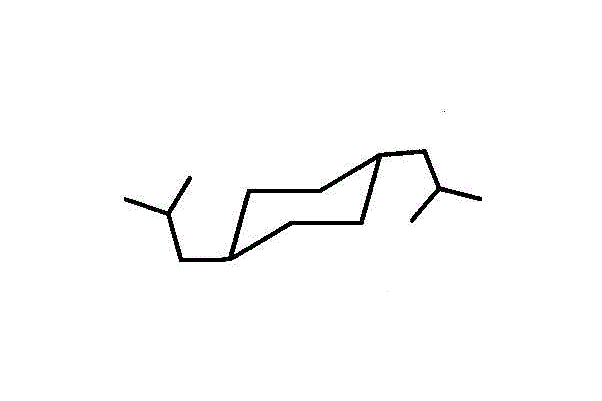
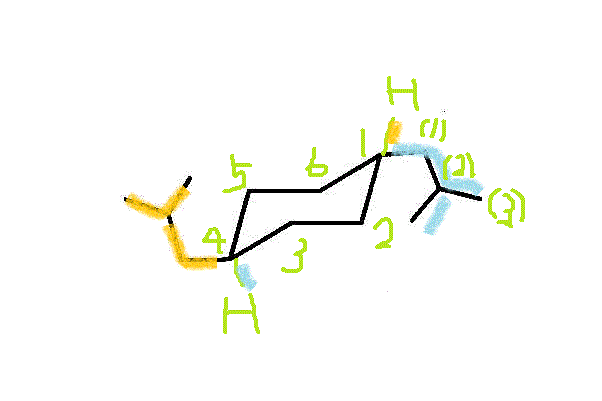
- 枝分かれの最長の鎖は3だから「プロピル基」、その根元から2番目に「メチル基」。こんな複雑な置換基が二つ同じであるときは、重複を避けて、bisとまとめる。・・・trans-1,4-bis(2-methylpropyl)cyclohexan
- 六角形の「面」の「上側」をオレンジ●、「下側」をブルー●で表すと、2つのメチル基は、「反対側」、だから「トランス」。・・・trans-1,2-dimethylcyclohexan
- 「橋かけ環式炭化水素」
- 2つの環があるからbicyclo-、炭素数は全部で7だからheptane。2つの環があるということは、枝分かれのポイント2点(橋頭炭素原子)から、3本の「橋」が架かっている、とも言える。各々の「橋」を構成する炭素数を大きいもの順に列挙すると、2個、2個、1個。こうして、・・・bicyclo[2,2,1]heptane
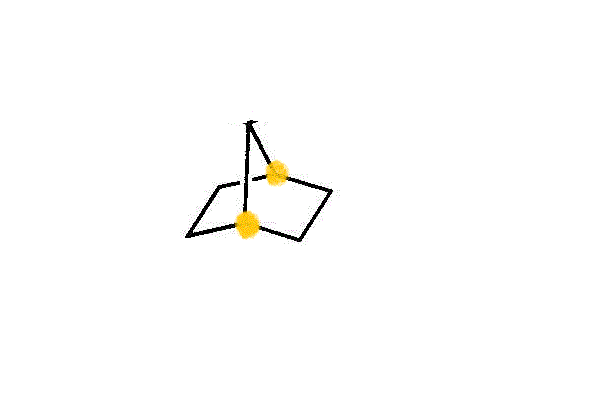
- 2つの環があるからbicyclo-、炭素数は全部で8だからoctane。各々の「橋」を構成する炭素数は、2個、2個、2個。・・・bicyclo[2,2,2]octane
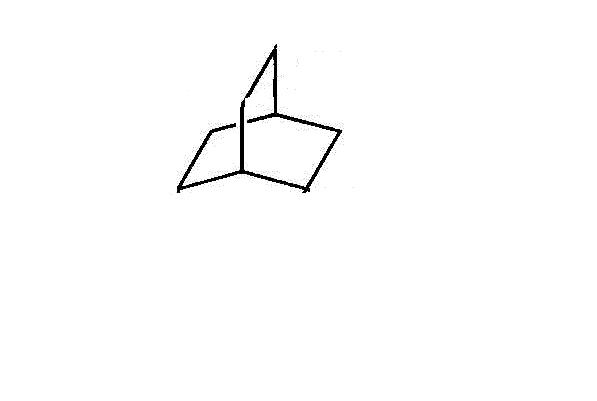
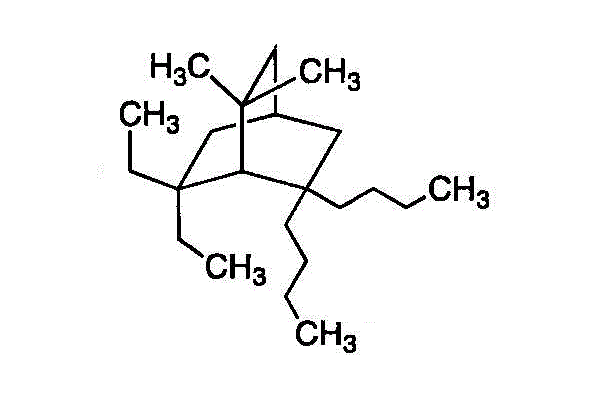
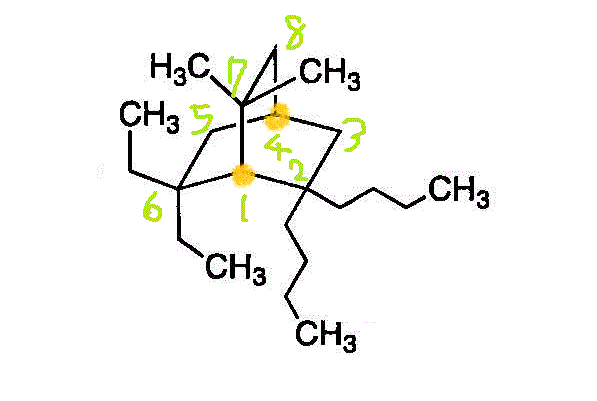
- 番号は、一方の「橋頭」から出発して、最長の橋を渡り、二番目に長い橋を戻ってくる。そのあとに、残りの橋を渡る。置換基の一番号が小さくなるようにすれば、手前の「橋頭」が1番、炭素数4の「ブチル基」が2個付いているのが、2番。橋の長さはどれも同じだが、付いている基がmethyl、ethyl、butylでアルファベット順を採用した。・・・2,2-butyl-6,6-ethyl-7,7-methylbicyclo[2,2,2]octane
- 1-methyl-7,7-dimethylbicyclo[2,2,1]heptane
(1,7,7-trimethylbicyclo[2,2,1]heptaneと思ったが、上の書き方がしてあるものがあった。)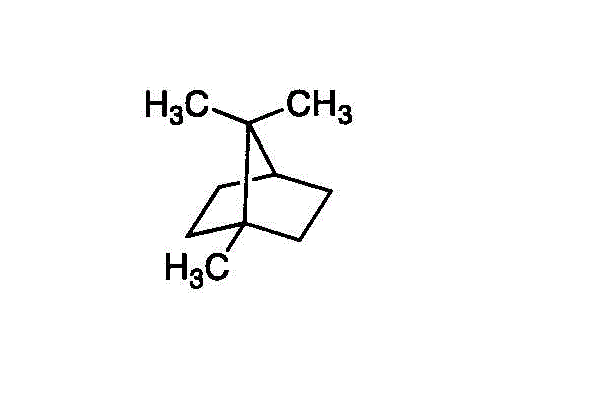
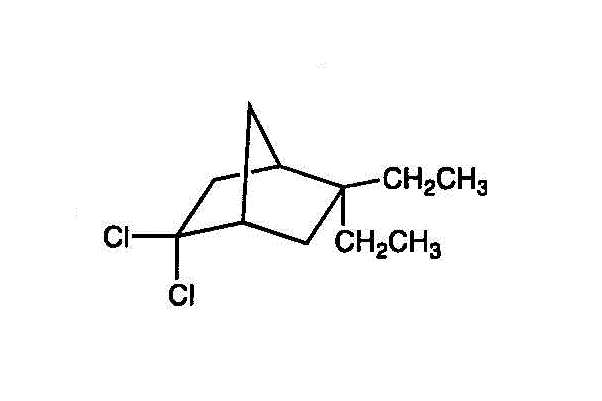
- 2,2-dichloro-5,5-diethylbicyclo[2,2,1]heptane
- 2,2-diethyl-7,7-diethyl-5,5-dimethylbicyclo[2,2,1]heptan
(これも、2,2,7,7-tetraethyl-5,5-dimethylbicyclo[2,2,1]heptaneではいけないのかどうかは、わからない。)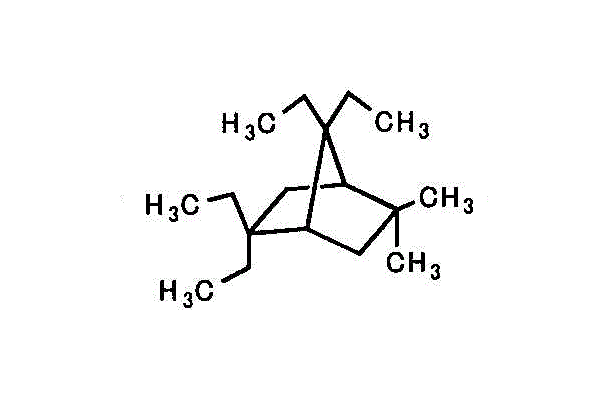
- 2つの環があるからbicyclo-、炭素数は全部で7だからheptane。2つの環があるということは、枝分かれのポイント2点(橋頭炭素原子)から、3本の「橋」が架かっている、とも言える。各々の「橋」を構成する炭素数を大きいもの順に列挙すると、2個、2個、1個。こうして、・・・bicyclo[2,2,1]heptane
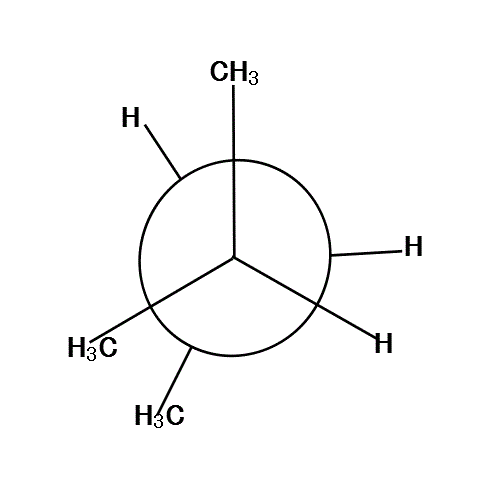
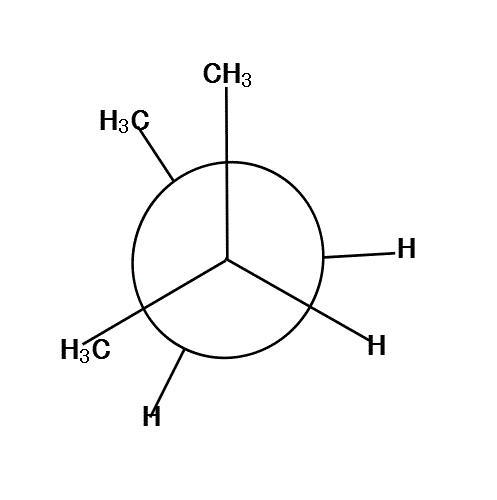
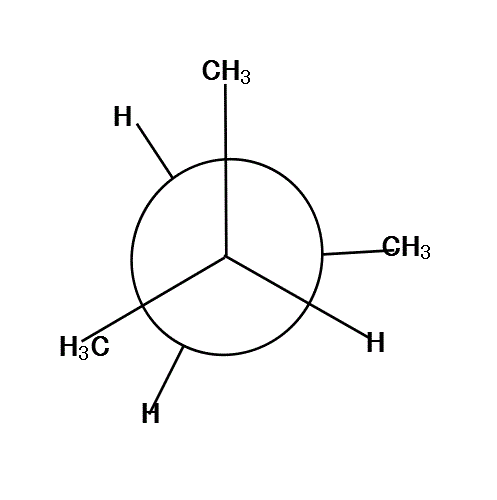
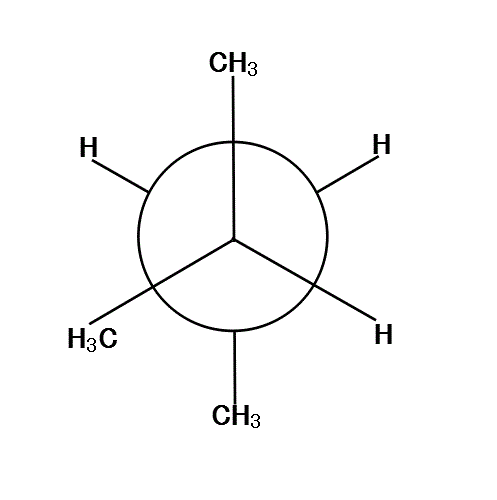
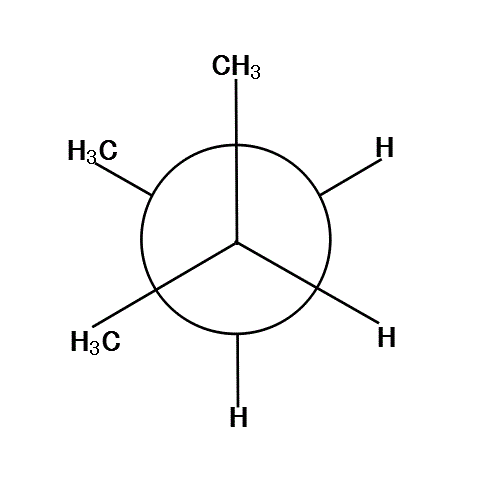
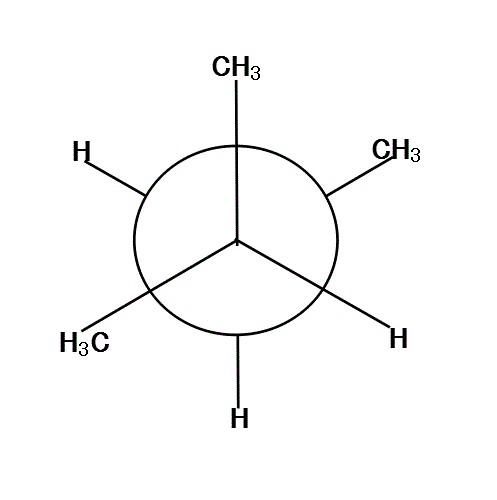
2-methylbutaneの、C2-C3結合を、C2側から覗き込む。「手前」のC2には2個の「メチル基」と「水素」が、ほぼ120°の角度をなして結合している。「奥」のC3には、1個の「メチル基」と2個の「水素」が結合している。
手前の方の「メチル基」を、真上と左下向きに固定して、「奥」の方を、「メチル基」が真下にある状態から順次、時計回りに回転していく。
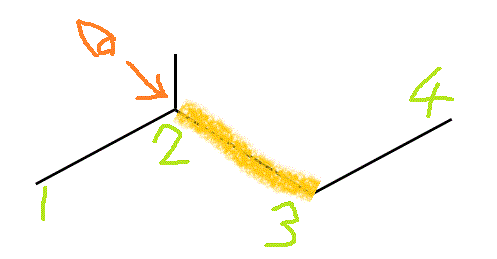
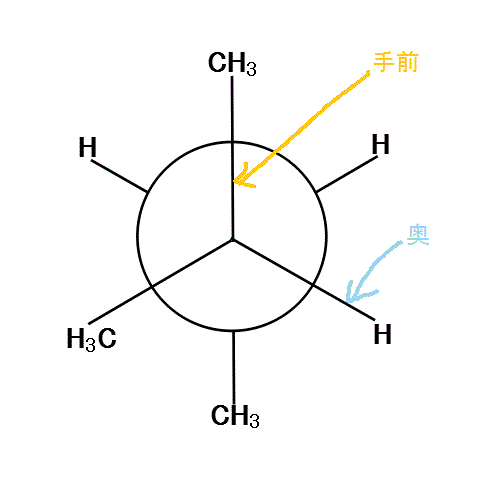
120°、240°、360°では、6本の腕が、ほぼ正六角形の頂点方向に分散しているから、安定である。
反対に、60°、180°、300°では、C2の基とC3の基が、重なり合って(図では重なると描けないから少しずらす)、不安定になる。
広い空間に、なるべく均等に広がったほうが「安定」で、局所的に混みあった状態は「不安定」なのである。この「不安定さ」を表示するのが、「ポテンシャル・エネルギー」、つまり「位置エネルギー」だが、これを外部に放出することで「安定化」する。
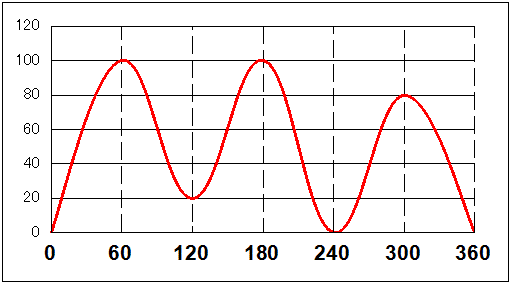
さらに細かく見ると、「混み合い方」は、基の大きさにも関係して、「水素」より「メチル基」の方がはるかに大きいから、
H⇔H<CH3⇔H<CH3⇔CH3
の順になっていくだろう。グラフで表すと、こんな感じ、もちろん縦軸の目盛は適当(!)である。
- メタンの塩素化
- 開始反応
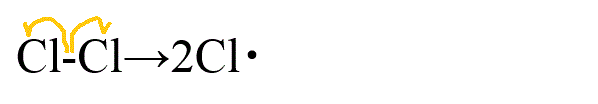
- 伝搬反応
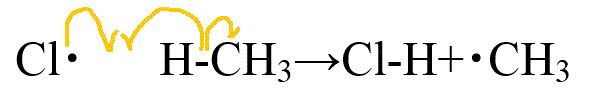
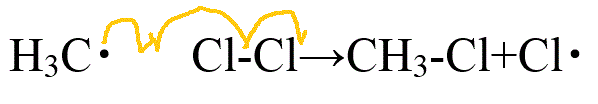
- 停止反応
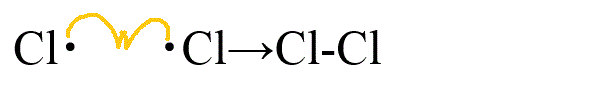
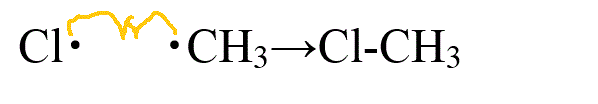
- 開始反応
- エタンの臭素化
- 開始反応

- 伝搬反応
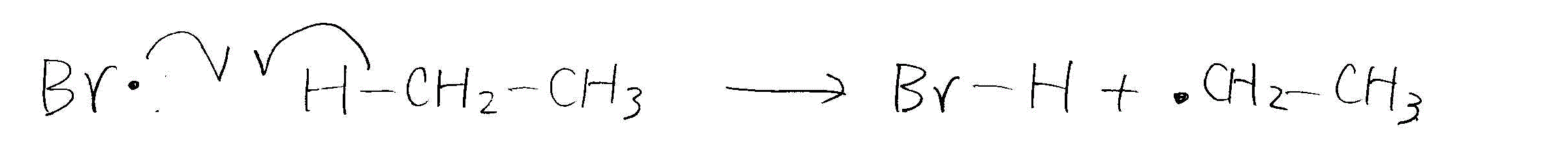
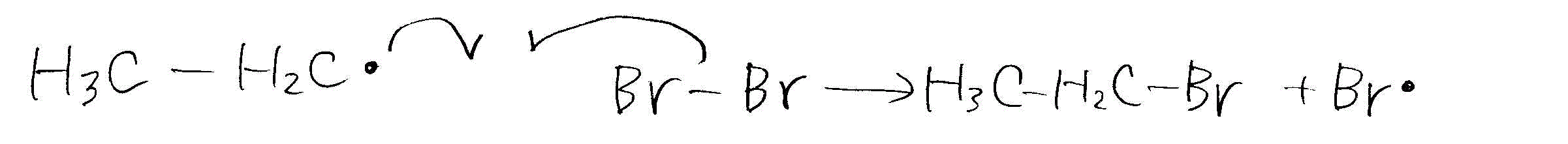
- 停止反応
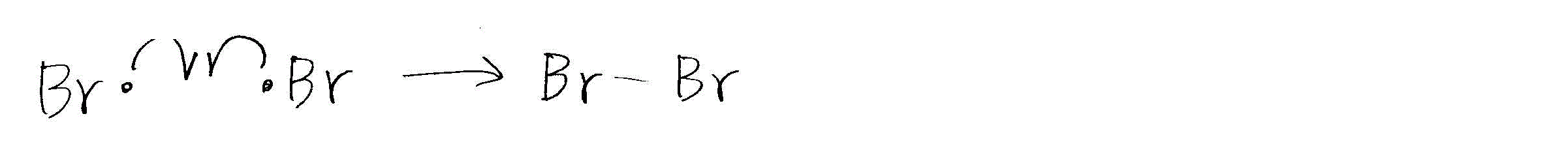
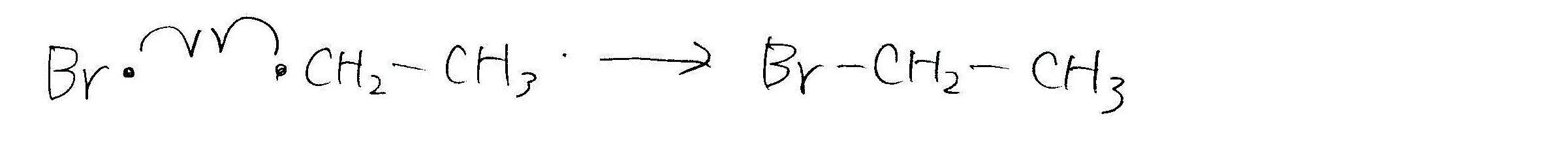
- 開始反応
- プロパンの塩素化
CH3-CH2-CH3+Cl2→CH3-CH2-CH2Cl+HCl(第1級水素の置換)
または、
CH3-CH2-CH3+Cl2→CH3-CHCl-CH3+HCl(第2級水素の置換)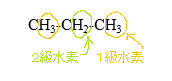
「第1級水素」は6個、「第2級水素」は2個あり、もし、その反応性が同じであるならば、その収量は、3:1の比率になるだろうところ、
実験の結果からは、43:57であった。これは、「第2級水素」の反応性が、「第1級水素」より、かなり高いことを意味している。どのくらい高いのか?
理論値と実測値の比から、57/1:43/3≒4:1、約4倍、反応性が高い。
(温度を高くすると、十分なエネルギーが与えられ、反応性の低い「第1級水素」もよく反応することになるから、収量の比は、理論値3:1に近づくだろう) - 2-メチルプロバンの塩素化
CH3-CH(CH3)-CH3+Cl2→CH3-CH(CH3)-CH2Cl+HCl(第1級水素の置換)
または、
CH3-CH(CH3)-CH3+Cl2→CH3-CCl(CH3)-CH3+HCl(第3級水素の置換)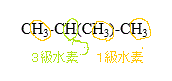
「第1級水素」は9個、「第3級水素」は1個あり、もし、その反応性が同じであるならば、その収量は、9:1の比率になるだろうところ、
実験の結果からは、64:36であった。これは、「第3級水素」の反応性が、「第1級水素」より、かなり高いことを意味している。どのくらい高いのか?
理論値と実測値の比から、36/1:64/9≒5:1、約5倍、反応性が高い。 - 2-メチルブタンの塩素化
以上2つの結果から、同じ温度条件のもとで、塩素に対しては、「第3級水素」、「第2級水素」、「第1級水素」の反応性は、5:4:1であると推測される。ここから、2-メチルブタンの塩素化によって生ずる化合物の収量の比を推定できる。- CH3-CH(CH3)-CH2-CH3+Cl2→CH2Cl-CH(CH3)-CH2-CH3+HCl(左端の第1級水素の置換)
- CH3-CH(CH3)-CH2-CH3+Cl2→CH3-CH(CH3)-CH2-CH2Cl+HCl(右端の第1級水素の置換)
- CH3-CH(CH3)-CH2-CH3+Cl2→CH3-CH(CH3)-CHCl-CH3+HCl(第2級水素の置換)
- CH3-CH(CH3)-CH2-CH3+Cl2→CH3-CCl(CH3)-CH2-CH3+HCl(第3級水素の置換)

生成物 水素の個数 反応性の比 収量の比 CH2Cl-CH(CH3)-CH2-CH3 6 1 6×1=6 CH3-CH(CH3)-CH2-CH2Cl 3 1 3×1=3 CH3-CH(CH3)-CHCl-CH3 2 4 2×4=8 CH3-CCl(CH3)-CH2-CH3 1 5 1×5=5 - 2-メチルプロパンのフッ素化/臭素化
F2 Cl2 Br2 反応性 高 ⇔ 低 選択性 低 ⇔ 高 - F2
- 反応性が高いから、遷移状態になるのが早い。反応物と、遷移状態の中間体の構造が似ている。
- 第1級水素の引き抜きと第3級水素の引き抜きのエネルギー差が小さい。選択性が低く、水素の個数の比(第1級:第3級)9:1に近くなる。
- Br2
- 反応性が低いから、遷移状態になるのが遅い。遷移状態の中間体の構造は、生成物と似ている。
- 第1級水素の引き抜きと第3級水素の引き抜きのエネルギー差が大きい。選択性が高い。よりエネルギーの低い第3級水素引き抜きの反応が起こりやすいため、水素の個数の比(第1級:第3級)9:1から大きく外れる。
- Br2に、Cl2を混ぜると?
- より選択性の低いCl2によって、エネルギーの高い第1級水素の引き抜きも進行するから、これに臭素ラジカルBr・が結合して第1級の生成物も生じることになり、Br2のみの場合より、選択性は低下する。
- F2
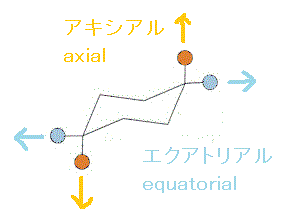
アキシアル位に大きな置換基が入ると、水素や他の置換基との距離が近く、立体的に「混み合う」から、不安定化する。
- cis-1,2-dimethylcyclohexan/trans-1,2-dimethylcyclohexan
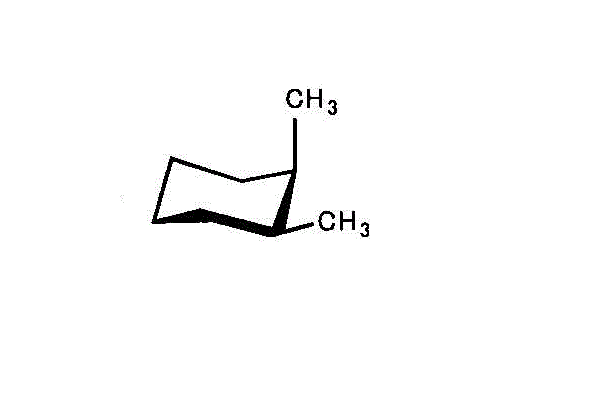
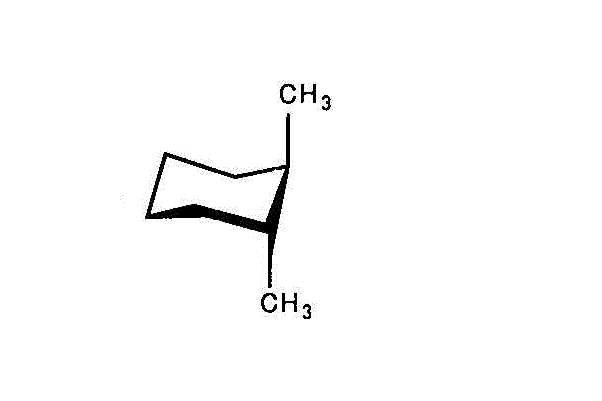
左「シス形」では、2つのメチル基のうち、一つはエクアトリアル、他はアキシアル。
右「トランス形」では、2つのメチル基いずれも、アキシアル。
アキシアルが多い「トランス型」の方が不安定で、エネルギーは高いだろう。 - cis-1,3-dimethylcyclohexan/trans-1,3-dimethylcyclohexan
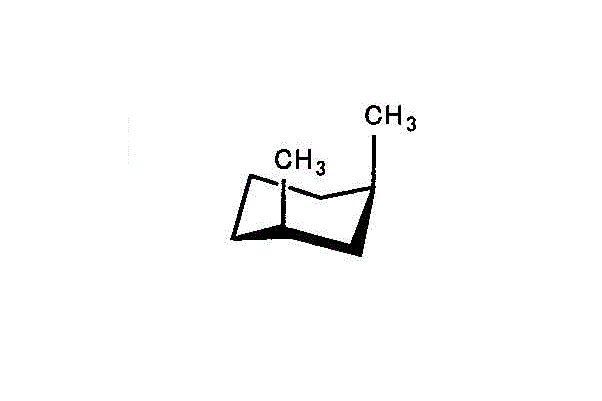
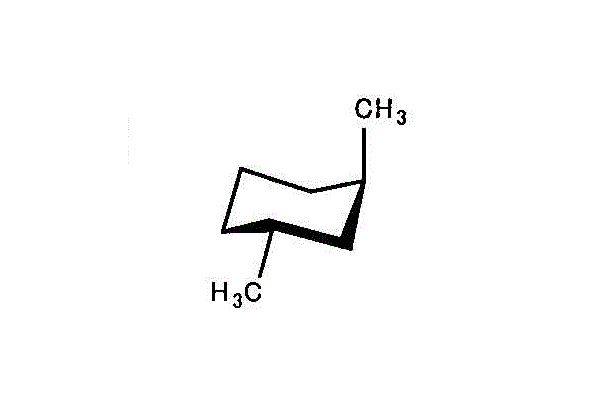
左「シス形」では、2つのメチル基いずれも、アキシアル。
右「トランス形」でも、2つのメチル基いずれも、アキシアル。
両者に、安定性の差はなく、エネルギーも等しいだろう。 - cis-1,4-dimethylcyclohexan/trans-1,4-dimethylcyclohexan
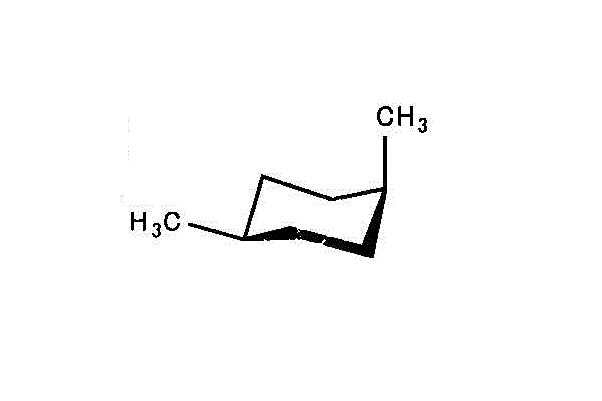
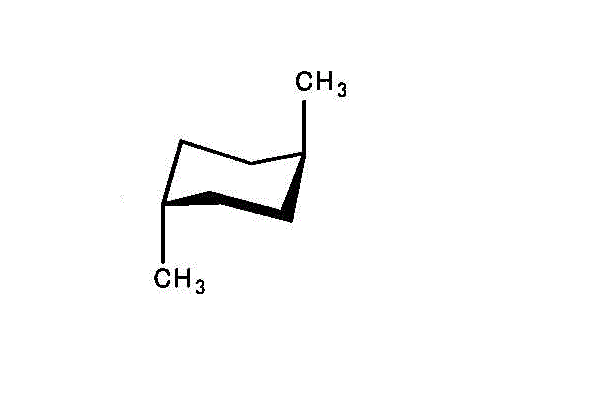
左「シス形」では、2つのメチル基のうち、一つはエクアトリアル、他はアキシアル。
右「トランス形」では、2つのメチル基いずれも、アキシアル。
アキシアルが多い「トランス型」の方が不安定で、エネルギーは高いだろう。